English
English Korean
KoreanDOMESTIC RA
ICMC (International Certification Management Center Co., Ltd.)
-
DOMESTIC RA
-
In Vitro Diagnostic KGMP

icmcert@naver.com

+82-2-851-3111
In Vitro Diagnostic KGMP
In vitro diagnostic medical device manufacturing and import GMP audit
In vitro diagnostic medical device GMP in Korea (MFDS)
In vitro diagnostic medical device GMP (Good Manufacturing Practice)
The in vitro diagnostic medical device GMP system refers to a quality guarantee system to ensure that in vitro diagnostic medical devices produced and imported are safe, effective, and consistently produced quality products suitable for intended use. Companies that want to produce and import in vitro diagnostic medical devices need to secure systematic quality systems for the entire process, such as designing and developing, manufacturing, and post-marketing management of in vitro diagnostic medical devices, and must obtain GMP approval.
In vitro diagnostic medical device GMP audit target
- Manufacturer or importer of in vitro diagnostic medical devices.
- Manufacturer or importer of in vitro diagnostic medical devices for clinical performance testing.
- Manufacturers or importers of in vitro diagnostic medical devices who wish to be recognized for suitability and undergo regular screening.
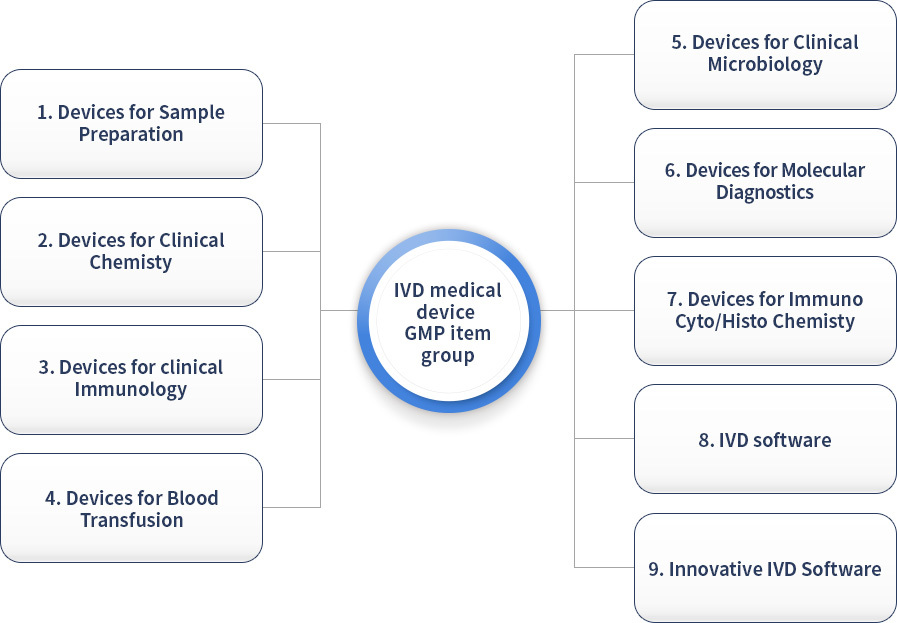
In vitro diagnostic medical device GMP audit item group
It is classified into nine GMP item groups according to the characteristics of each item, and GMP suitability is recognized for each item group. (Additional review is required when adding item groups)
In vitro diagnostic medical device GMP audit classification
- 1
- Initial audit
In vitro diagnostic medical devices manufactured or imported for the first time to be recognized as meeting GMP standards.
- 2
- Regular audit
Regular examination of GMP application performance that must be received once every three years in accordance with the Enforcement Rules of the In Vitro Diagnostic Medical Devices Act and GMP Notice (application for regular examination 90 days before the expiration of the validity period of the appropriate certificate).
- 3
- Additional audit
Examination received in cases where in vitro diagnostic medical devices of other item groups are to be manufactured or imported pursuant to attached Table 3 of the GMP Notice.
- 4
- Change audit
Examination received when the location of the manufacturer is changed (transferred) (excluding changes in storage and laboratory that are less related to the quality of the product).
In vitro diagnostic medical device GMP audit method
In vitro diagnostic medical device GMP audit subject (exclusive: examination agency, joint: local food agency/examination agency)
| Classification | Manufacturing industry | Importing industry |
|---|---|---|
Class 2
| Exclusive examination (one person from the examination agency) | Exclusive examination (one person from the examination agency) |
| Class 3, 4 | Joint examination (1 or 2 persons from the Regional Food and Drug Administration, 1 person from the examination agency) | Joint examination (1 or 2 persons from the Regional Food and Drug Administration, 1 person from the examination agency) |
| Audit period | Calculation of the number of days of examination (MD) according to the number of employees (2-7 days) | Calculation (4-7 days) in consideration of the complexity of the manufacturing process and the business trip schedule (local situation, etc.) |
In vitro diagnostic medical device GMP audit method
| Classification | Manufacturing industry | Importing industry |
|---|---|---|
Initial audit
| On-site audit, document audit | On-site audit, document audit |
| Additional audit | Document audit | Document audit |
| Change audit | On-site audit, document audit | On-site audit, document audit |
| Regular audit | On-site audit, document audit (In vitro diagnostic medical devices of Class 1 are excluded from regular audits) |
On-site audit, document audit (In vitro diagnostic medical devices of Class 1 are excluded from regular audits) |
Application for in vitro diagnosis medical device GMP audit
In vitro diagnostic medical device GMP audit application
Submit the required documents to the examination agency in an application for audit, such as recognition of suitability, etc.
- Application for audit, such as in vitro diagnostic medical device suitability recognition, etc.
- A copy of the in vitro diagnostic medical device manufacturing (import) business license or a copy of the conditional manufacturing (import) business license (excluding in the case of in vitro diagnostic medical devices for initial examination or clinical performance testing).
- Data necessary for the audit, such as conformity recognition, etc.
- A copy of the audit result notice, such as in vitro diagnostic medical device technical document, etc. (in the case of in vitro diagnostic medical devices for clinical performance testing, technical documents, etc.)
※ Quality control performance of at least one manufacturing unit is required for representative items (with a completed product test report).
In vitro diagnostic medical device GMP audit procedure
In vitro diagnostic medical device GMP audit procedure

In vitro diagnostic medical device GMP evaluation criteria
-
Evaluation table evaluation criteriaA
Where compliance with the GMP standard requirements is recognized. BIf the GMP standard requirements are not followed,
If the GMP standard requirements are observed, but the basis for proof, feasibility, and records of compliance are insufficient,CIn the case of not supplementing "B (Requirement for Supplement)" or violating the Medical Device Act, DIf it does not meet the requirements of the GMP criteria -
Criteria for determining suitabilityConformity
If all items are appropriate (A) as a result of the audit by audit criteria supplementationAs a result of the audit for each audit standard, if one or more items need to be supplemented (B) NonconformityIf one or more items are inappropriate (C) as a result of the audit by audit criteria
In vitro diagnostic medical device GMP requirements
In vitro diagnostic medical device GMP process

In vitro diagnostic medical device GMP requirements
| Classification | Detailed requirements | Classification | Detailed requirements |
|---|---|---|---|
4. Quality management system
| 4.1 General requirements |
7. Product realization | 7.1 Planning of product realization |
| 5. Management responsibility | 5.1 Management commitment |
8. Measurement, analysis and improvement | 8.1 General |
| 6. Resource management | 6.1 Provision of resources |
Application of GMP monitoring standards for suitability of in vitro diagnostic medical devices
Usability refers to design features (e.g., buttons, user screens, user manuals, etc.) designed to reduce the risk of use errors that may occur when using a product and allow users to accurately and safely use in vitro diagnostic medical devices.
GMP usability application is implemented step by step for each in vitro diagnostic medical device grade, and quality control techniques that apply usability when designing and developing in vitro diagnostic medical devices should be specifically presented and applied to in vitro diagnostic medical device manufacturers.
In vitro diagnostic medical device usability design and evaluation consist of steps such as (1) preparation of specifications for use, (2) safety check of user interface, (3) preparation and selection of related scenarios for predictable hazards, (5) establishment of user interface specifications, and (7) general evaluation of user interface usability.
From January 1, 2021 to July 1, 2022, it will be implemented in stages by grade.
| Class | Class 4 | Class 3 | Class 2 | Class 1 |
|---|---|---|---|---|
Effective date
| 2021. 1. 1 |
2021. 7. 1 |
2022. 1. 1 |
2022. 7. 1 |
Introduction of in vitro diagnostic medical device standard code (UDI) system
Manufacturers and importers of in vitro diagnostic medical devices must mark standard codes (number indicated according to standardized systems, bar codes including electronic tags, etc.) on the container or exterior of in vitro diagnostic medical devices.
From July 1, 2019 to July 1, 2022, it will be implemented in stages by grade.
| Class | Class 4 | Class 3 | Class 2 | Class 1 |
|---|---|---|---|---|
Effective date
| 2019. 7. 1 |
2020. 7. 1 |
2021. 7. 1 |
2022. 7. 1 |
Post-management of in vitro diagnostic medical devices
Report on in vitro diagnostic medical device foreign substance
Manufacturing, import, and repair companies of in vitro diagnostic medical devices must prepare and submit the previous year's production, export, import, and repair performance once a year from January 1st to January 31st every year.
Report on the supply of in vitro diagnostic medical devices
In vitro diagnostic medical devices manufacturing, importing, selling, and leasing companies must submit a "supply breakdown report" to the Minister of Food and Drug Safety through the Integrated Medical Device Information System" by the end of the following month.
From July 1, 2020 to July 1, 2023, it will be implemented in stages by grade.
| Class | Class 4 | Class 3 | Class 2 | Class 1 |
|---|---|---|---|---|
Effective date
| 2020. 7. 1 |
2021. 7. 1 |
2022. 7. 1 |
2023. 7. 1 |
In vitro diagnostic medical device
If an in vitro diagnostic medical device handler finds a substance (foreign substance) that may cause harm or is inappropriate to use when used as it is not a raw material used normally inside, containers, or packaging the in vitro diagnostic medical device, he/she shall immediately submit a Foreign Object Discovery Report to the Ministry of Food and Drug Safety.
In vitro diagnostic medical device abnormality report
If an in vitro diagnostic medical device handler recognizes that death or serious side effects may occur or occur while using the in vitro diagnostic medical device, he/she must submit the Medical Device Abnormal Case Report to the Minister of Food and Drug Safety from the date he/she knows it to the next day.
| The subject of reporting | The deadline for reporting | |
|---|---|---|
In vitro diagnostic medical device handler. (In vitro diagnostic medical device manufacturer, import, repair, sales, rental business, medical institution founder, animal hospital founder) |
Within 7 days |
- In the case of death or life-threatening abnormalities, ※ Additional details are reported within 8 days from the date of initial report. |
Within 15 days |
- In the case where it is necessary to extend the hospitalization or hospitalization period, - In the event that recovery is impossible, serious disability, or deterioration of function is caused. - In the case of congenital malformations or abnormalities, |
|
Within 30 days |
- In the case where the head of the Ministry of Food and Drug Safety ordered a report as other serious information or other abnormal cases, - In the case of measures such as announcement by foreign governments, ※ However, if the recovery plan is reported, it can be omitted. |
|
Medical personnel, patients, in vitro diagnostic medical device consumers. |
- In the case of learning a strange case | |
In vitro diagnostic medical device GMP consulting stage in Korea (MFDS)

Related major websites
- Company : ICMC (International Certification Management Center Co., Ltd.)
- CEO : Bong Ju Kim
- Address: 16 Digital-ro 32-ga-gil, Guro-gu, Seoul (Guro-dong) Partner Tower 2nd Room 1001.
- TEL : +82-2-851-3111
- FAX : +82-2-2028-3115
- BIZ NO : 214-88-14891
- EMAIL : icmcert@naver.com
 10004 certified
10004 certified- 27701 Certified
- Copyright(c) ICMC International Certification Management Center. All Rights Reserved.