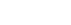English
English Korean
KoreanDOMESTIC RA
ICMC (International Certification Management Center Co., Ltd.)
-
DOMESTIC RA
-
In Vitro Diagnostic(Import/ Export)

icmcert@naver.com

+82-2-851-3111
In Vitro Diagnostic(Import/ Export)
In vitro diagnostic medical device law of Korea (MFDS)
The In vitro Diagnostic Medical Device Act in Korea stipulates the definition and permission of in vitro diagnostic medical devices, and is subdivided into enforcement ordinances and enforcement rules and contains the following details.
Definition of in vitro diagnostic medical devices in Korea (MFDS)
Medical devices under Article 2 (1) of the Medical Devices Act, such as reagents, contrast and correction materials, equipment, machinery, devices, software, etc., used alone or in combination to examine samples derived from humans or animals in vitro (Article 2 of the In Vitro Diagnostic Medical Devices Act)
- A product used to diagnose physiological or pathological conditions.
- A product used for the purpose of determining the predisposition of a disease or observing the prognosis of a disease.
- A product used to provide information on congenital disabilities.
- A product used for the purpose of providing information necessary for safety and suitability judgment when transfusing or transplanting blood, tissue, etc. to another person.
- Products used for the purpose of predicting treatment response and treatment outcome
- Products used for the purpose of determining treatment methods or monitoring treatment effects or side effects.
Classification of in vitro diagnostic medical devices in Korea (MFDS)
In vitro diagnostic medical devices are classified into four classes according to the purpose of use and the degree of potential risk to personal and public health.
* Regulations on in vitro diagnostic medical device items and classes by item (Notice No. 2020-34).
- Class 1 - Reagents with low potential risks to individuals and public health, and devices used for legal diagnostic purposes.
- Class 2 - Devices with potential risk of severity to individuals but low potential risk to public health
- Class 3 - Devices used for tests that have a decisive effect on diagnosis, treatment, disease stage determination, and treatment.
- Class 4 - A test to select donors for transfusion or transplantation to others or a device used when personal risk is high.
In vitro diagnostic medical device item classification in Korea (MFDS)
| Classification of in vitro diagnostic medical devices (Middle Classification by Large Classification) | |||
|---|---|---|---|
1. (I) Devices for Sample Preparation |
2. (J) Devices for Clinical Chemistry |
3. (K) Devices for Clinical Immunology |
4. (L) Devices for Blood Transfusion |
- Centrifuge for medical use |
- Clinical chemistry analyzer |
- Clinical immune response analyzer |
- Blood transfusion analyzer |
5. (M) |
6. (N) |
7. (O) |
8. (P) |
- Clinical microbiology analyzer |
- Molecular diagnostic analyzer
|
- Cell/Histopathology test analyzer |
- IVD software for diagnosis |
9. Innovative IVD Software
|
|||
Korea (MFDS) In vitro diagnostic medical device licensing system
Before the market.
As manufacturers, pre-marketing regulators are managed by dividing them into analytical performance tests, clinical performance tests, item reporting, certification and permission, and GMP suitability recognition, a manufacturing system manager.
After selling it,
Post-marketing regulations are under follow-up management through systems such as advertising pre-deliberation systems, side effects/safety reports, and re-evaluation and re-examination.
In vitro diagnostic medical device licensing procedure in Korea (MFDS)
Class 1 : Item report (Korea Medical Device Safety Information Service, NIDS)
- - Application for item report [In vitro diagnostic medical device electronic complaint counter] → Registration after registration is completed.
- Contents including technical documents: name (product name, item name, model name) / classification number (class) / shape and structure / purpose of use / method of use / precautions for use / Manufacturer (in case of import or manufacturing process consignment) / identity product license number
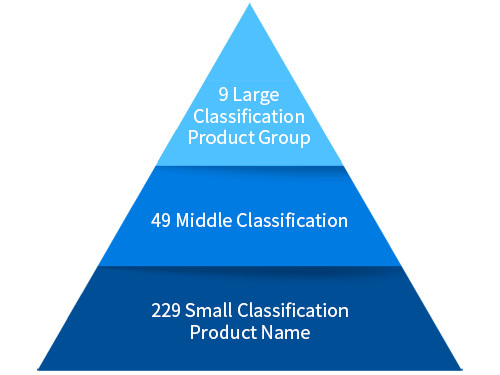
Class 2 : Item certification (Provided by the Ministry of Food and Drug Safety)
- Item classification criteria
- Equivalent product classification criteria: Purpose of use, principle of operation, raw material, performance.
- New product: In vitro diagnostic medical devices that have already been approved, and in vitro diagnostic medical devices that do not have the same purpose of use and principle of action.
- Improved product: In vitro diagnostic medical devices that have already been approved, and in vitro diagnostic medical devices that have the same purpose and operation principle but do not have equal raw materials or performance.
- Equivalent product: In vitro diagnostic medical devices that have already been approved, and in vitro diagnostic medical devices with equal purposes, principles of operation, raw materials, and performance.
Class 3-4 : Item permission (consideration and progress at the headquarters of the Ministry of Food and Drug Safety)
- Class 2 and flow are the same. The review period for technical documents is 65 days and 80 days for collective application for permission, and there is a difference in the period.
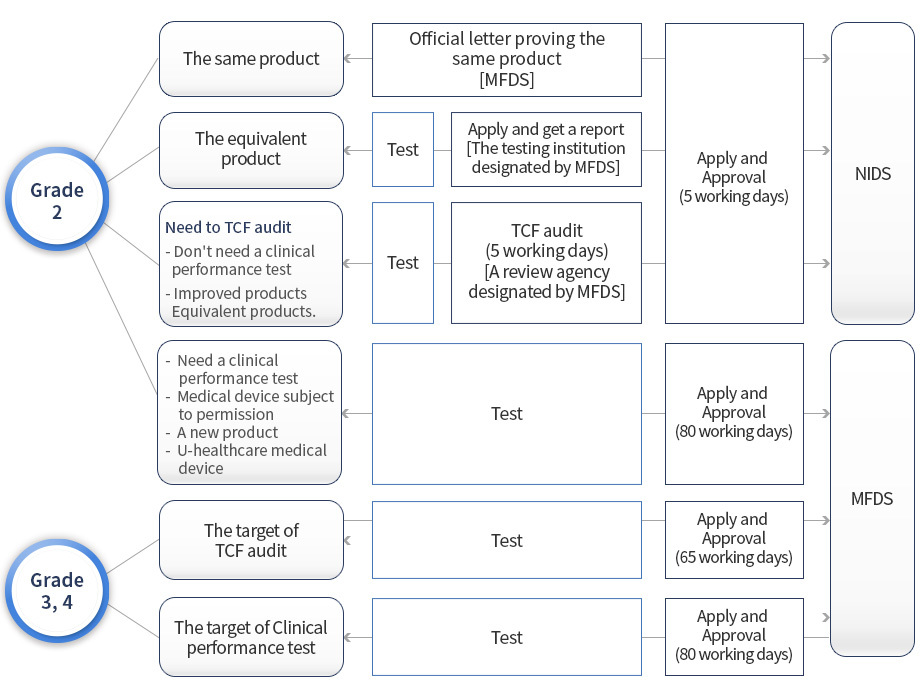
Certification of products subject to comparison of class 2 essential equivalent items
In vitro diagnostic medical device GMP in Korea (MFDS)
In vitro diagnostic medical device GMP stipulates the requirements of the quality management system applied in designing, developing, producing, installing, and providing services for medical devices.
Detailed GMP requirements can be found in the In Vitro Diagnostic Medical Device Manufacturing and Quality Control Standards Notice.
It applies to all in vitro diagnostic medical device manufacturers and importers, and the types of screening are divided into document screening and on-site screening.
After the initial examination, you must undergo a regular renewal examination every three years. If additional items other than the medium-sized items classified by GMP occur, there may be additional on-site inspections if the location is changed.
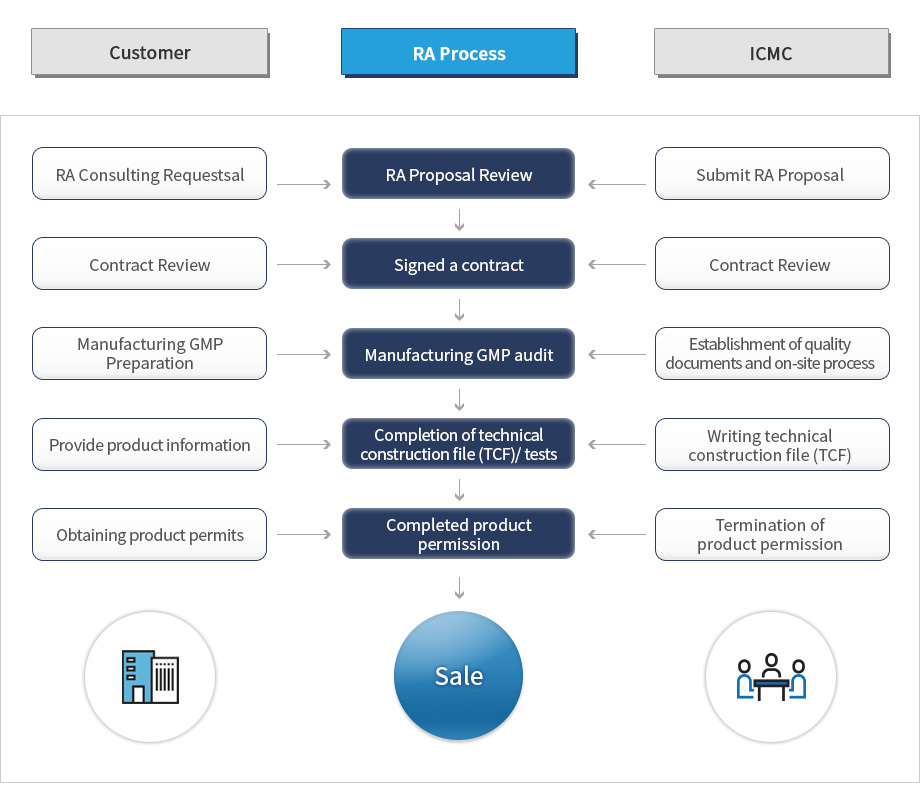
In vitro diagnostic medical device RA consulting stage in Korea (MFDS)
Related major websites
- Company : ICMC (International Certification Management Center Co., Ltd.)
- CEO : Bong Ju Kim
- Address: 16 Digital-ro 32-ga-gil, Guro-gu, Seoul (Guro-dong) Partner Tower 2nd Room 1001.
- TEL : +82-2-851-3111
- FAX : +82-2-2028-3115
- BIZ NO : 214-88-14891
- EMAIL : icmcert@naver.com
 10004 certified
10004 certified- 27701 Certified
- Copyright(c) ICMC International Certification Management Center. All Rights Reserved.